
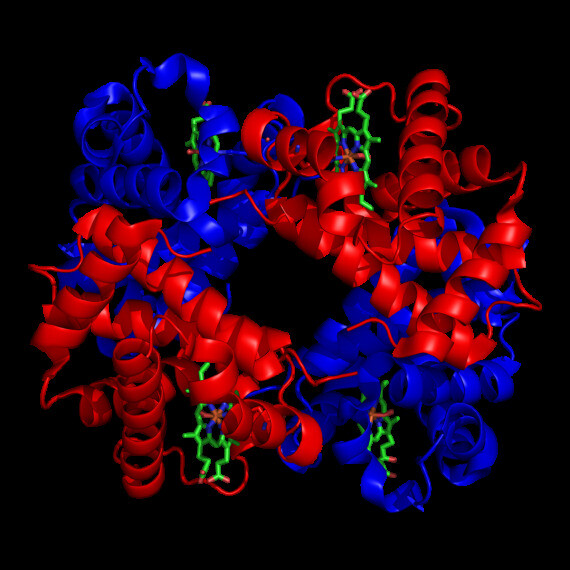
 Cấu trúc phân tử hemoglobin. Nguồn ảnh: Wikipedia.com Hemoglobin (Hb) là một đại phân tử có trong tất cả các tế bào hồng cầu. Nó cấu tạo từ 4 phân đơn vị gồm hem và globin. Hem là sắc tố chứa sắt có chức năng liên kết với oxy. Globin là một chuỗi protein được ký hiệu là alpha, beta, gamma hoặc delta. Các phân tử protein này liên kết với nhân hem ở trung tâm.
Cấu trúc phân tử hemoglobin. Nguồn ảnh: Wikipedia.com Hemoglobin (Hb) là một đại phân tử có trong tất cả các tế bào hồng cầu. Nó cấu tạo từ 4 phân đơn vị gồm hem và globin. Hem là sắc tố chứa sắt có chức năng liên kết với oxy. Globin là một chuỗi protein được ký hiệu là alpha, beta, gamma hoặc delta. Các phân tử protein này liên kết với nhân hem ở trung tâm.
Không phải tất cả hemoglobin đều giống nhau. Chúng được phân loại theo loại chuỗi globin thành phần.
Các loại hemoglobin bình thường bao gồm:
- Hemoglobin A: đây là loại Hb chủ yếu ở người lớn (chiếm 95-98%). Hb A chứa hai chuỗi protein alpha (α) và hai chuỗi protein beta (ß).
- Hb A2: chiếm khoảng 2-3,5% Hb ở người lớn. Nó có hai chuỗi protein alpha (α) và hai chuỗi delta (δ).
- Hb F: chiếm 2% Hb ở người lớn. Nó có hai chuỗi protein alpha (α) và hai gamma (γ). Hb F được sản xuất chủ yếu trong thời kì bào thai, sau đó giảm dần và về mức thấp trong vòng một năm sau khi sinh.
Video FBNC - Bệnh Thalassemia - nguyên nhân và cách điều trị
Những người mắc bệnh thalassemia có một hoặc nhiều đột biến di truyền làm giảm sản xuất hemoglobin. Khi lượng hem lành mạnh không đảm bảo, nó có thể ảnh hưởng đến quá trình cung cấp oxy cho cơ thể. Từ đó, bạn xuất hiện các triệu chứng của bệnh từ nhẹ đến nặng như suy nhược, mệt mỏi và da xanh.
Có bốn gen trong DNA quy định tổng hợp alpha globin và hai gen cho mỗi chuỗi beta, delta và gamma globin. Các dạng đột biến cấu trúc này có thể di truyền từ cha mẹ sang con cái. Chúng có thể làm giảm sản xuất, đảo lộn sự cân bằng của các chuỗi globin và hình thành hemoglobin bất thường. Thalassemia thường được phân loại dựa theo chuỗi globin bị ảnh hưởng. Càng nhiều gen bị đột biến, triệu chứng của bệnh càng nặng.
Một số đột biến gen mã hóa chuỗi globin có thể làm thay đổi cấu trúc globin dẫn đến môt số bệnh lý như hồng cầu hình liềm.
Alpha thalssemia
 Phân tích ADN cho phép xác định người mang gen đột biến thầm lặng. Nguồn ảnh: Generalelectric.com
Phân tích ADN cho phép xác định người mang gen đột biến thầm lặng. Nguồn ảnh: Generalelectric.comBệnh alpha thalassemia gây ra bởi sự mất đoạn hoặc đột biến ở các gen quy định cho alpha globin. Càng nhiều gen bị ảnh hưởng, cơ thể càng sản xuất ít globin này hơn. Có bốn loại alpha thalassemia khác nhau được phân loại theo số lượng gen bị ảnh hưởng:
- Người mang mầm bệnh thầm lặng (1 gen bị ảnh hưởng). Những người này không xuất hiện triệu chứng của bệnh và thường chỉ được chẩn đoán khi sinh con mắc bệnh thalassemia. Cách duy nhất để xác định người mang gen đột biến thầm lặng là phân tích DNA. Xét nghiệm máu cho thấy nồng độ hemoglobin và các chỉ số hồng cầu bình thường.
- Thalassemia nhẹ (2 gen bị ảnh hưởng). Những người này có các tế bào hồng cầu nhỏ hơn, giảm nồng độ hemoglobin trung bình trong hồng cầu (MCHC), giảm huyết sắc tố trung bình hồng cầu (MCH) và thiếu máu nhẹ. Họ thường không hoặc chỉ xuất hiện một số ít triệu chứng. Dạng thiếu máu này không đáp ứng với việc bổ sung sắt. Chẩn đoán thường được dựa trên phương pháp loại trừ các nguyên nhân thiếu máu hoặc phân tích DNA.
- Bệnh Hemoglobin H (3 gen bị ảnh hưởng). Sự sụt giảm lớn trong sản xuất chuỗi alpha globin gây dư thừa chuỗi beta, tạo thành các phân tử Hemoglobin H chứa 4 chuỗi beta, có thể nhìn thấy được trong tế bào hồng cầu trên lam máu. Bệnh Hb H có thể gây thiếu máu từ trung bình đến nặng với các biến chứng nghiêm trọng như lách to, biến dạng xương, thiếu máu nghiêm trọng.
- Phù thai nhi (4 gen bị ảnh hưởng). Đây là dạng alpha thalassemia nghiêm trọng nhất. Trong trường hợp này, thai nhi không thể sản xuất được alpha globin và xuất hiện các đặc điểm lâm sàng như gan lách to, khuyết tật tim, tràn dịch não. Bệnh thường được chẩn đoán nhờ siêu âm trong những tháng cuối thai kì. Trong một số trường hợp, mẹ cũng bị ảnh hưởng với các triệu chứng như protein niệu, tăng huyết áp, phù chân và xuất huyết nặng sau sinh. Thai nhi thường bị sẩy, chết lưu hoặc chết ngay sau khi sinh. Trong một số trường hợp rất hiếm, trẻ có thể sống sót nhờ được truyền máu trong tử cung và chăm sóc đặc biệt.
Alpha thalassemia thường gặp ở những người gốc Đông Nam Á, Nam Trung Quốc, Trung Đông, Ấn Độ, Châu Phi và Địa Trung Hải.
Beta thalassemia
 Hồng cầu nhỏ bất thường là một dấu hiệu của thalassemia. Nguồn ảnh: Azolifesciences.com
Hồng cầu nhỏ bất thường là một dấu hiệu của thalassemia. Nguồn ảnh: Azolifesciences.comBệnh beta thalassemia là do đột biến ở một hoặc cả hai gen quy định tổng hợp beta globin. Đã có hơn 250 đột biến được xác định, nhưng chỉ có khoảng 20 trong đó thường xảy ra. Mức độ nghiêm trọng của bệnh phụ thuộc vào loại đột biến và mức độ giảm sản xuất beta globin. Các thể bệnh beta thalassemia bao gồm:
- Beta Thalassemia nhẹ. Những người bệnh này có một gen bình thường và một gen bị đột biến làm giảm nhẹ sản xuất beta globin. Họ thường không biểu hiện triệu chứng bệnh ra bên ngoài. Xét nghiệm cho thấy tế bào hồng cầu nhỏ bất thường và không đáp ứng với thuốc bổ sung sắt. Các gen đột biến này có thể di truyền từ bố mẹ sang con.
- Beta thalassemia trung bình. Trong trường hợp này, người bệnh có hai gen bất thường gây giảm sản xuất beta globin. Mức độ nghiêm trọng của bệnh phụ thuộc vào mức độ thiếu tần suất truyền máu cần thiết. trong một số trường hợp, bệnh nhân không cần truyền máu thường xuyên.
- Thalassemia thể nặng hoặc Thiếu máu Cooley. Đây là dạng bệnh beta thalassemia nặng nhất. Người bệnh mang trong mình hai gen bất thường gây giảm nghiêm trọng hoặc thiếu hụt hoàn toàn beta globin, từ đó ngăn cản tổng hợp một lượng đáng kể hemoglobin bình thường (Hb A). Tình trạng thiếu máu này thường xuất hiện trong hai năm đầu đời và đe dọa đến tính mạng, làm giảm tăng trưởng và hình thành các bất thường xương sơ sinh. Thể bệnh này đòi hỏi truyền máu thường xuyên suốt đời và được theo dõi bởi các bác sĩ. Tuy nhiên, truyền máu thường xuyên này dẫn đến tích tụ sắt trong các cơ quan như gan, tim. Nếu không được điều trị, người bệnh có thể tử vong sớm do suy đa tạng. Do đó, trong phác đồ điều trị cần được bổ sung các phương pháp thải sắt.
Beta thalassemia được tìm thấy phổ biến trong các quần thể người Mỹ gốc Địa Trung Hải, châu Phi, và Đông Nam Á.
Một số dạng thalassemia khác xảy ra khi gen bệnh beta thalassemia được di truyền kết hợp với gen của một hemoglobin bất thường. Chúng bao gồm:
- Hb E-beta thalassemia. Hb E là một trong những đột biến hemoglobin phổ biến. Nó được tìm thấy chủ yếu ở những người gốc Đông Nam Á và châu Phi. Nếu một người có cả gen đột biến tổng hợp Hb E và beta thalassemia thì chúng có thể kết hợp tạo ra Hb E-beta thalassemia, một bệnh lý gây thiếu máu tương tự như bệnh beta thalassemia thể trung bình.
- Hb S-beta thalassemia hoặc beta thalassemia hồng cầu hình liềm. Hb S là một trong những đột biến hemoglobin được biết đến nhiều nhất. Người mắc Hb S-beta thalassemia có cả gen Hb S và gen beta thalassemia. Mức độ nghiêm trọng của bệnh phụ thuộc vào lượng beta globin được sản xuất.
Xét nghiệm cận lâm sàng
 Phết máu ngoại biên giúp đánh giá số lượng và hình dạng của tế bào bạch cầu, hồng cầu và tiểu cầu. Nguồn ảnh: Agric.wa.gov.au
Phết máu ngoại biên giúp đánh giá số lượng và hình dạng của tế bào bạch cầu, hồng cầu và tiểu cầu. Nguồn ảnh: Agric.wa.gov.auMột số xét nghiệm sau có thể giúp phát hiện và chẩn đoán thalassemia:
Công thức máu cơ bản (CBC - Complete blood count). Xét nghiệm này giúp đánh giá các chỉ số như số lượng hồng cầu, kích thước và hình dạng hồng cầu và thể tích hồng cầu trung bình (MCV). MCV thấp là triệu chứng của thalassemia và thiếu máu thiếu sắt.
Phết máu ngoại biên. Đây là kỹ thuật được thực hiện để kiểm tra một mẫu máu ngoại biên bằng kính hiển vi. Cụ thể, bác sĩ sẽ lấy một ít máu của người bệnh lên tấm kính và quan sát tiêu bản dưới kính hiển vi. Xét nghiệm này đánh giá số lượng và hình dạng của tế bào bạch cầu, hồng cầu và tiểu cầu. Với bệnh thalassemia, hồng cầu có thể:
- Nhỏ hơn bình thường
- Giảm huyết sắc tố
- Thay đổi về kích thước và hình dạng
- Có nhân (hồng cầu trưởng thành không có nhân)
- Phân bố hemoglobin không đồng đều
Tỷ lệ hồng cầu bất thường càng lớn thì khả năng mang oxy càng kém cũng như nguy cơ mắc bệnh lý về máu càng cao.
Xét nghiệm sắt huyết thanh. Chúng bao gồm: lượng sắt, ferritin, khả năng liên kết sắt không bão hòa (UIBC), tổng khả năng liên kết sắt (TIBC) và độ bão hòa transferrin. Các kết quả cho thấy khả năng dự trữ và sử dụng sắt của cơ thể. Chúng được chỉ định để xác định xem thiếu sắt có phải là nguyên nhân gây thiếu máu ở bệnh nhân hay không. Chúng cũng được chỉ định để xác định mức độ dư thừa sắt ở người đang điều trị thalassemia.
- Alpha thalassemia đôi khi bị nhầm lẫn với thiếu máuthiếu sắt vì chúng đều biểu hiện bởi các tế bào hồng cầu nhỏ hơn bình thường. Tuy nhiên, người bệnh thalassemia không có nồng độ sắt trong máu thấp. Bổ sung sắt sẽ không mang lại hiệu quả với người mắc alpha thalassemia và có thể dẫn đến tình trạng thừa sắt, gây tổn thương lên các cơ quan nội tạng.
- Xét nghiệm porphyrin máu có thể sử dụng để phân biệt beta thalassemia thể nhẹ với thiếu sắt hoặc ngộ độc chì. Người mắc beta thalassemia sẽ có mức porphyrin bình thường, trong khi các bệnh còn lại có nồng độ porphyrin trong máu cao.
 Điện di huyết sắc tố giúp đánh giá thành phần và tỷ lệ hemoglobin trong máu. Nguồn ảnh: Healthcheckup.com
Điện di huyết sắc tố giúp đánh giá thành phần và tỷ lệ hemoglobin trong máu. Nguồn ảnh: Healthcheckup.comĐiện di huyết sắc tố. Xét nghiệm này đánh giá thành phần và tỷ lệ hemoglobin trong máu. Hemoglobin A (Hb A) có thành phần gồm cả alpha và beta globin, là loại hemoglobin chủ yếu chiếm 95% đến 98% hemoglobin ở người lớn. Hemoglobin A2 (HbA2) thường chiếm từ 2% đến 3%, trong khi hemoglobin F thường chiếm dưới 2%.
Bệnh beta thalassemia làm mất cân bằng chuỗi beta và alpha globin, qua đó làm thay đổi tỉ lệ thành phần hemoglobin. Vì vậy, người mắc bệnh beta thalassemia thể nặng thường tăng tỷ lệ Hb F, người mắc thể nhẹ có tỷ lệ Hb A2 cao hơn. Hb H là một dạng hemoglobin ít phổ biến hơn có thể gặp trong một số trường hợp alpha thalassemia. Hb S là hemoglobin ít phổ biến và xuất hiện ở người bị bệnh hồng cầu hình liềm.
Điện di huyết sắc tố (Hb) được sử dụng để sàng lọc và đánh giá nguy cơ bất thường huyết sắc tố của thai nhi trước sinh.
Phân tích ADN. Các xét nghiệm này được sử dụng để xác nhận các đột biến trong gen sản xuất alpha và beta globin, quá đó giúp chẩn đoán thalassemia và xác định người mang gen bệnh.
- Với người nghi ngờ mắc beta thalassemia, gen beta hemoglobin (HBB) có thể được phân tích hoặc giải trình tự để xác nhận sự hiện diện của các đột biến gây bệnh. Hơn 250 đột biến có liên quan đến bệnh beta thalassemia và một số trong chúng không gây ra bất kì triệu chứng nào. Tuy nhiên, nếu chúng làm giảm sản xuất beta globin, bác sĩ có thể chẩn đoán xác định beta thalassemia.
- Xét nghiệm sinh học phân tử cũng giúp phát hiện các đột biến phổ biến có trong gen alpha HBA1 và HBA2. Chúng có thể là nguyên nhân gây bệnh alpha thalassemia.
Nếu phát hiện người thân mắc bệnh này, những người khác cũng nên xét nghiệm để xác định có gen đột biến hay không.
Xét nghiệm nước ối cũng được thực hiện nếu cả cha và mẹ đều mang đột biến vì sự kết hợp các gen bất thường có thể gây ra một thể thalassemia nghiêm trọng ở thai nhi.
Điều trị
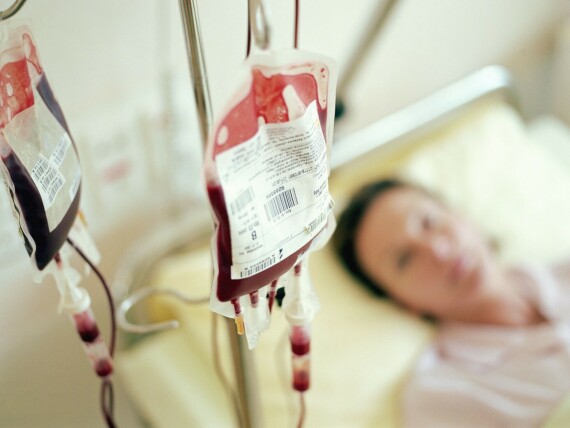 Người bệnh thalassemia có thể cần đến truyền máu để duy trì đủ lượng hemoglobin. Nguồn ảnh: Nih.gov
Người bệnh thalassemia có thể cần đến truyền máu để duy trì đủ lượng hemoglobin. Nguồn ảnh: Nih.govHầu hết những người mắc bệnh thalassemia thể nhẹ đều không cần điều trị. Tuy nhiên, họ nên được tư vấn di truyền vì các gen đột biến có thể truyền lại cho con cái.
Những người bị bệnh hemoglobin H hoặc beta thalassemia thể trung bình sẽ có những giai đoạn thiếu máu khác nhau. Họ có thể sống một cuộc sống tương đối bình thường nhưng đôi khi có thể cần truyền máu. Bổ sung acid folic có thể giúp cải thiện tình trạng bệnh, tuy nhiên, họ không nên sử dụng thêm sắt.
Những người mắc bệnh beta thalassemia thể nặng sẽ phải truyền máu thường xuyên, vài tuần một lần kết hợp với liệu pháp thải sắt trong suốt cuộc đời. Việc truyền máu giúp duy trì hemoglobin ở mức đủ cao để cung cấp oxy cho cơ thể và ngăn ngừa các bất thường về tăng trưởng cũng như tổn thương các cơ quan. Tuy nhiên, việc truyền máu thường xuyên có thể làm tích tụ sắt trong các cơ quan đến mức gây độc. Do đó, cơ thể cần được thải sắt định kì.
Ghép tủy xương hay ghép tế bào gốc tạo máu cũng có thể được sử dụng để điều trị bệnh nhân beta thalassemia thể nặng.
Thai nhi mắc bệnh alpha thalassemia thể nặng thường bị sẩy, chết lưu hoặc chết ngay sau khi sinh. Một số phương pháp như truyền máu cho thai nhi và thậm chí cấy ghép tủy cho thai nhi đã được thực hiện thành công trong một số rất ít trường hợp.
Xem thêm: